Agammaglobulinemia: X-linked (XLA) and autosomal recessive (ARA)
People with agammaglobulinemia can't produce antibodies, which are an important part of the body's defense against germs.
The more you understand about primary immunodeficiency (PI), the better you can live with the disease or support others in your life with PI. Learn more about PI, including the various diagnoses and treatment options.

Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.

Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.

Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.

People with agammaglobulinemia can't produce antibodies, which are an important part of the body's defense against germs.
Some people with primary immunodeficiency (PI) lack the cells that are responsible for producing antibodies (immunoglobulins). They have very low or absent levels of all types of antibodies and an increased risk of infection. People with genetic causes of low or absent antibodies (agammaglobulinemia) have a severe form of antibody deficiency with absent B cells. This results from the failure of precursor B cells to develop into mature B cells and plasma cells.
The basic defect in agammaglobulinemia is the inability of the patient to produce antibodies. Antibodies are an integral part of the body's defense mechanism against germs. When a germ, such as bacteria, lands on a mucous membrane or enters the body, antibody molecules that recognize the germ stick to its surface.
Antibodies bound to the surface of a germ can have one or more effects that are beneficial. For example, some germs must attach to body cells before they can cause an infection and antibodies prevent the germs from sticking to the body's cells. Antibody on the surface of some germs will also activate other body defenses, such as a group of blood proteins called serum complement, which can directly kill the bacteria or viruses. Finally, antibody-coated bacteria are much easier for white blood cells (phagocytes) to ingest and kill than bacteria that are not coated with antibody.
All of these actions prevent germs from invading body tissues where they may cause serious infections. Antibodies are also important in the recovery from infections and to protect against getting certain infections more than once.
Antibodies are produced by specialized cells in the body, called plasma cells. Plasma cells are derived from B lymphocytes (a type of white cell) that develop in an orderly sequence of steps beginning with stem cells located in the bone marrow. The stem cells give rise to immature lymphocytes that eventually develop into mature lymphocytes including T cells, B cells, or NK cells. A subset of these immature lymphocytes called pro-B lymphocytes next develop into pre-B lymphocytes, which then give rise to mature B lymphocytes (Figure 2:1).
.png")
Each B lymphocyte has antibodies on its cell surface. Each B cell makes a slightly different antibody, or immunoglobulin, to allow the body to respond to millions of different foreign substances (also known as antigens), like vaccines or parts of bacteria or viruses. There are specific antibodies designed to recognize each unique antigen. When the B lymphocyte comes into contact with its antigen, it is triggered to mature into a plasma cell. Plasma cells specialize in making and secreting large amounts of specific antibodies.
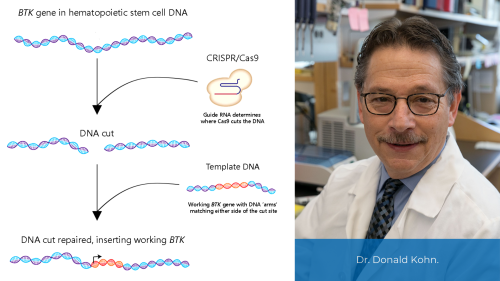
The first form of agammaglobulinemia to be recognized, X-Linked agammaglobulinemia (XLA), was described in 1952 by Colonel Ogden Bruton, MD. This disease, sometimes called Bruton’s agammaglobulinemia or congenital agammaglobulinemia, typically affecting boys, was the first type of PI to be identified. Most individuals with XLA have normal numbers of B cell precursors, but very few of these are able to go on to become mature B cells. People with XLA have variants, or changes, in a gene that is necessary for the normal development of B cells. This gene, discovered in 1993, is named Bruton’s tyrosine kinase (BTK) in honor of Dr. Bruton.
After BTK variants were identified as the cause of XLA, it became clear that only about 85% of children with agammaglobulinemia and absent B cells had variants in BTK. In addition, it had been known for several years that there were girls who had an immunodeficiency that looked just like XLA, with agammaglobulinemia and absent B cells, but which could not be explained by X-linked inheritance of BTK variants.
Since 1996, several genes that can cause agammaglobulinemia with autosomal recessive inheritance (ARA) have been identified. The following genes (and their official gene symbol) have been reported to cause ARA:
In addition, more recently, autosomal dominant forms of agammaglobulinemia have been described due to variants in LRRC8 (LRRC8A) and E2A (TCF3).
All of these genes code for proteins involved in the maturation of B cells. Individuals with variants in any of these genes have clinical and laboratory findings that are very similar to those seen in those with variants in BTK.
See if you qualify to participate in clinical trials evaluating new treatments and/or diagnostics for agammaglobulinemia.
The diagnosis of agammaglobulinemia should be considered in any individual (male or female) with recurrent or severe bacterial infections, particularly if they have small or absent tonsils and lymph nodes.
The first screening test should be an evaluation of serum immunoglobulins. In most individuals with agammaglobulinemia, all of the immunoglobulins (IgG, IgM, IgA, IgE) are low or absent. In addition, healthy babies make only small quantities of immunoglobulins, particularly IgA and IgE, in the first few months of life, making it difficult to distinguish a healthy baby with a delay in immunoglobulin production from a baby with true immunodeficiency.
If the serum immunoglobulins are low or if the healthcare provider strongly suspects the diagnosis of agammaglobulinemia, the number of B cells in the blood should be measured. A low percentage of B cells (1% or less of the lymphocytes) in the blood is the most characteristic and reliable laboratory finding in someone with agammaglobulinemia.
If a newborn baby has a parent, sibling, maternal cousin, or maternal uncle with agammaglobulinemia, the baby is at risk to have a similar immunodeficiency, and the family and healthcare providers should immediately determine the percentage of B cells in the blood so that treatment can be started before an affected infant gets sick.
The diagnosis of XLA can be confirmed by demonstrating the absence of BTK protein in monocytes or platelets or by the detection of a variant in the BTK gene. Almost every family has a different variant in BTK; members of the same family, however, usually have the same mutation. The specific gene that causes ARA can be identified by genetic testing.
Antibody deficiency with absent B cells are a group of genetic diseases, and can be inherited or passed on in a family. It is important to know the type of inheritance so the family can better understand why a child has been affected, the risk that subsequent children may be affected, and the implications for other members of the family.
As the name XLA suggests, the BTK gene (which is mutated in XLA) is located on the X chromosome. Since XLA is an X-linked disorder, typically only boys are affected because they have only one X chromosome (XY). Girls can be carriers of the disorder because they have two X chromosomes. Carriers of XLA typically have no symptoms, but they have a 50% chance of transmitting the disease to each of their sons.
Now that the precise gene that causes XLA has been identified, it is possible to test the female siblings (sisters) of a male with XLA, and other female relatives, such as the child’s maternal aunts, to determine if they are carriers of the disease and could transmit it to their sons. It is also possible to determine if a fetus of a carrier female will be born with XLA.
ARA occurs when an individual inherits two copies of a gene (typically one from each parent), each of which has a variant that makes the gene not function. This is more likely when parents are related in some way to one another or come from a small, isolated geographic region or close-knit community. In autosomal recessive forms of agammaglobulinemia, an individual who inherits only one gene with a variant and has one functional gene will not be affected. Individuals with autosomal dominant forms of agammaglobulinemia need to only inherit one copy of the gene variant to be affected.
Most individuals with antibody deficiency with absent B cells, who receive Ig replacement therapy on a regular basis, will be able to lead relatively normal lives. They do not need to be isolated or limited in their activities. Children with agammaglobulinemia can participate in all regular school and extracurricular activities, and active participation in team sports should be encouraged. Infections may require some extra attention from time to time, but many with antibody deficiency with absent B cells can go on to become adults with productive careers and families. A full active lifestyle is to be encouraged and expected.

XLA Life fosters the unification and empowerment of the global X-Linked Agammaglobulinemia (XLA) community through education, advocacy, and initiatives that aim to improve the overall quality of life for those affected by XLA.
This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice. Additionally, links to other resources and websites are shared for informational purposes only and should not be considered an endorsement by the Immune Deficiency Foundation.
Adapted from the IDF Patient & Family Handbook for Primary Immunodeficiency Diseases, Sixth Edition.
Copyright ©2019 by Immune Deficiency Foundation, USA
Receive news and helpful resources to your cell phone or inbox. You can change or cancel your subscription at any time.